
Innate Immunity and Bacterial Pathogenesis
All living things must defend themselves against infection — this is the job of the immune system. In the Vance Lab, we focus on the initial events that occur upon the first encounter between a host and a pathogen. In particular, we are interested in answering three fundamental questions: (1) how does the innate immune system detect pathogens? (2) how are pathogen infections distinguished from benign encounters with harmless microbes? and (3) how does the innate immune response contribute to the elimination of pathogens in the face of pathogen virulence strategies?
We study a diversity of different infection models and use a variety of experimental techniques, from structural biology to biochemistry to genetics to in vivo studies. Despite our varied experimental approaches, several themes recur in our studies. For example, we focus primarily on bacterial infections. We are also particularly interested in pathogen sensors that are situated in the cytosol of host cells, since we hypothesize these sensors are preferentially activated by pathogens (versus non-pathogens). And lastly, we are interested in how the immune system detects pathogen-encoded activities, what we have termed “patterns of pathogenesis“.
Currently the lab is working in several areas:
Inflammasomes: guardians of the host cell cytosol
Inflammasomes are multi-protein complexes that assemble in the cytosol of host cells upon detection of various noxious or infectious stimuli. Once assembled, inflammasomes serve as a scaffold for the dimerization and activation of inflammatory caspases, most notably, Caspase-1. Cells express several different inflammasomes, each of which is responsive to distinct stimuli. The Vance Lab has focused primarily on two inflammasomes: the NAIP–NLRC4 inflammasome that detects bacterial proteins such as flagellin; and the NLRP1 inflammasome that detects pathogen-derived toxins and enzymes.
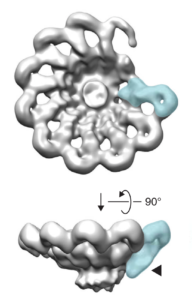
NAIP5/NLRC4: In collaboration with the Nogales Lab on campus, we solved the cryo-EM structure of the NAIP/NLRC4 inflammasome bound to flagellin (pictured above). Our paper provides insights into how flagellin is detected and why pathogens cannot easily mutate flagellin to escape detection. We have also recently studied the function of the NAIP5/NLRC4 inflammasome in defense against Salmonella and Shigella infection, in auto-inflammatory disease, and in induction of adaptive immunity.
NLRP1: The NLRP1 inflammasome provides resistance against infection with Bacillus anthracis, the causative agent of anthrax, but the mechanism by which NLRP1 is activated has been mysterious. We showed that proteolytic cleavage of NLRP1 results in its activation, and discovered the highly surprising “functional degradation” mechanism by which this occurs.
In our ongoing work, we want to better understand roles of inflammasomes during infection, as well as how pathogens circumvent detection by inflammasomes. In particular, our interest in inflammasomes has led to ongoing work in the lab on Shigella flexneri, the bacterial pathogen that causes dysentery (see below).
Cytosolic sensing of cyclic-di-nucleotides and nucleic acids
A series of fortuitous observations led us to discover that bacterial molecules called cyclic-di-nucleotides stimulate a specific innate immune response characterized by the production of type I interferons. By using random chemical mutagenesis of mice, we discovered that a host protein called STING is essential for the cytosolic interferon response to cyclic-di-nucleotides. We further showed that STING is a direct cytosolic receptor for cyclic-di-nucleotides. It was subsequently shown by the Chen lab that cyclic-di-nucleotides are not unique to bacteria but are also produced by a cytosolic DNA sensor protein called cGAS. We then showed that the cyclic-di-nucleotides produced by cGAS have a unique chemical structure that allows them to potently stimulate human STING.
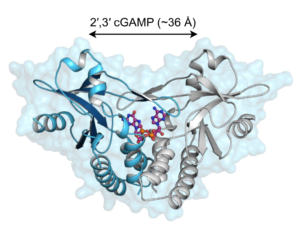
We have recently become particularly interested in the evolutionary origins of the STING protein. We were surprised to discover that even sea anemone, organisms that diverged from humans more than 500 million years ago, encode a functional STING protein that binds cyclic-di-nucleotides (pictured above). Sea anemone lack type I interferons, so an important question we are currently pursuing is: what is the evolutionarily ancient function of STING? and is this function conserved in mammalian STING? We have pursued this by laboratory studies on the sea anemone Nematostella vectensis. In separate by related work, we recently made the surprising discovery that interferon-independent activities of STING contribute to protection against HSV-1 in vivo.
Legionella pneumophila: detection of pathogen-encoded activities
L. pneumophila is a bacterium that is the causative agent of a severe pneumonia called Legionnaires’ Disease. We are interested in how the innate immune system detects L. pneumophila. In fact, our initial studies on L. pneumophila were what led us to identify the NAIP/NLRC4 inflammasome as an important sensor of flagellin. Our work on L. pneumophila also led to our interest in cyclic-di-nucleotides. More recently, we showed that additional innate sensing pathways also contribute to immune recognition of L. pneumophila. For example, we discovered an unusual immune response that is triggered upon blockade of host protein synthesis by L. pneumophila. This response appears to be critical for the initial immune response to L. pneumophila in the lung. We used ribosome profiling to show that this response depends upon a dramatic transcriptional superinduction of specific alarm signals called cytokines. One of our recent papers demonstrated that several L. pneumophila effectors modulate host cell metabolism, and in particular, affect the function of the master metabolic regulator mTOR—presumably this is all part of the strategy L. pnuemophila uses to obtain nutrients from host cells.
Mycobacterium tuberculosis: novel factors controlling lung inflammation
Recently, we have become particularly interested in applying our knowledge of innate immunity to an important globally significant pathogen. Mycobacterium tuberculosis, the causative agent of tuberculosis, is the single pathogen responsible for more human deaths than any other viral, bacterial or protozoan pathogen. M. tuberculosis was discovered by Robert Koch in 1882, but we remain largely ignorant about fundamental aspects of its microbiology and immunopathogenesis. Our current studies on M. tuberculosis were stimulated by a sabbatical visit from Heran Darwin. In addition, we are lucky to have two great colleagues at Berkeley, Jeff Cox and Sarah Stanley, who have helped us cope with the technical challenges of working with this BSL3 organism. We are emplying mouse genetic approaches to identify critical host factors that mediate susceptibility and resistance to M. tuberculosis. By taking advantage of a mouse model developed by our collaborator Igor Kramnik at Boston University, we discovered how a type I interferon response elicited by M. tuberculosis actually benefits the pathogen. We have also discovered that a novel transcriptional regulator called SP140 is an important mediator of immune resistance to M. tuberculosis. We are using our newly generated Sp140–/– mice to better understand the pathogenesis of this globally important disease. We recently used our Sp140–/– mice to identify the cellular sources and targets of the type I interferons that appear to drive TB pathogenesis.
Shigella flexneri: new approaches to a globally significant intestinal pathogen
Shigella is a genus of bacteria responsible for severe gastrointestinal infections—shigellosis and dysentery—in humans. It is estimated that there are upwards of 200,000 annual deaths each year due to Shigella, many of which occur in children under the age of five, primarily in developing countries. A major impediment to research on Shigella has been the lack of an in vivo mouse model that faithfully recapitulates the hallmarks of human disease. Our work on inflammasomes has led us to develop the first oral infection mouse model of shigellosis. We are now positioned to answer many fundamental questions about Shigella pathogenesis in vivo, with the aim of exploiting this knowledge in the design of improved vaccines and therapeutics. Our most recent work has suggested that cell death of epithelial cells is one critical host defense mechanism against Shigella.
Effector-triggered immunity
A nascent area of interest for the lab is the discovery of novel pathways that are activated in response to the virulence activities (or “effectors”) of pathogens. This kind of immune response is sometimes called “effector-triggered immunity” and is distinct from the much more commonly studied PAMP-triggered immunity. We discovered a surprising new pathway, called the MORC3 pathway, that is activated by an important virulence factor of HSV-1. When activated, the MORC3 pathway leads to the induction of potent anti-viral cytokines called type I interferons. Surprisingly, though, the canonical signaling pathways that are well-known to induce type I interferons do not seem to employed by the MORC3 pathway. Instead, a novel DNA element appears to be activated in the MORC3 pathway, leading to potent transcription of the interferon beta gene. We speculate that this novel pathway may have evolved as a countermeasure against viral effectors that block canonical interferon signaling. We are continuing efforts to discover additional examples of effector-triggered immunity.